آموزش بسته محاسباتی سیستا ( SIESTA )
سیستا یا SIESTA یک کد محاسبات کوانتومی است که توانایی انجام محاسبات ابتدا به ساکن الکترونیکی (ab initio) و محاسبات دینامیک مولکولی (molecular dynamics) را دارد. سیستا این کار را به بهینهترین شکل ممکن از نظر زمانی انجام میدهد. که این هم ناشی از مجموعهی توابع پایهی بسیار جایگزیده ای است که استفاده میکند. علاوه بر اینها، در سیستا از الگوریتمهای خطی سازی بسیار پیشرفتهای استفاده میشود. بهینه بودن این الگوریتم ها در حدی است که ناشرین ادعا میکنند با افزایش حجم یاخته، زمان محاسبات فقط دو برابر میشود و نه هشت برابر. این دقیقا برخلاف آن چیزی است که در کدهای تمام الکترونی که از رهیافت های دوگانه (والانس = امواج تخت + مغزه= مجموعه توابع اتمی ) یا کدهای مبتنی بر شبه پتانسیل که عمدتاً از پایه های تخت استفاده میشود، میبینیم. چون در این دو رهیافت کافیست اندکی حجم ابریاخته را افزایش دهید؛ زمان محاسبات با توان های عجیب و غریب افزایش مییابد. ولی سیستا برای محاسبات کلاستری و انواع حالتهای نانو ساخته شده است.
در این دوره مقدماتی، بیشتر هدف ما درک نحوه نوشتن ورودی ها و یافتن بهینه پارامترهای محاسباتی است. با این آموزش قادر خواهید بود دست کم پروژه ی دوران ارشد یا دکترایتان را به انتها برسانید. سعی میشود مثالهایی که در اینجا ارائه میدهیم، همه کارهای ابتدایی را شامل شود. با ثبت نام در این دوره میتوانید:
۱- نحوه نوشتن فایل ورودی ، بهینه کردن پارامترهای محاسباتی و واهلش ساختاری را بیاموزید.
۲- انواع مختلف ساختارها مثل حالت انبوهه ، انواع حالتهای نانو و کلاستر را بسازید و یک اجرای خودسازگار روی آنها انجام دهید.
۳- ساختار نواری ، چگالی حالت های الکترونی ( DOS )، محاسبات مغناطیسی و بیشتر کارهای پس پردازشی را محاسبه و رسم کنید.
با توجه به درخواست شما و تعداد افرادی که در این دوره ثبت نام میکنند، ممکن است مولفههای دیگر آموزشی نیز اضافه شوند.
در این جلسه خلاصه ای از کارهایی که در بیشتر کدهای کوانتومی انجام میشود، ارائه شده است. بعلاوه پارامترهایی که باید بهینه شوند نیز در این جلسه معرفی شده اند که در جلسه آینده آنها را بهینه خواهیم کرد.
در این جلسه کلیات مربوط به نوشتن فایلهای ورودی در کد محاسباتی سیستا توضیح داده شده است. همچنین kgrid و MeshCutOff نیز با نوشتن یک اسکریپت ساده، بهینه شدند.
یک نکته در انتخاب kpoint ها:
اگر ساختار 0-بعدی مثل کلاستر دارید، تعداد kpoint ها را برابر یک، ساختار یک بعدی مثل نانو ریبون در صورتی که راستای بینهایت آن در جهت x است، kpointها را k 1 1 بدهید.
برای ساختار دو و سه بعدی نیز k1 k2 1 و k1 k2 k3 مقداردهی کنید.
دلیل انتخاب اعداد 1 کوچک بودن منطقه اول بریلوئن و عدم پاشندگی در جهت های گفته است. با توجه به اینکه سیستا فقط تقارن وارونی زمانی را در نظر میگیرد، پس تعداد kpoint ها در بهترین حالت نصف خواهد شد. و اینکه سیستا نسبت به kpoint ها انعطاف زیادی ندارد. پس در انتخاب آنها دقت کنید.
MeshCutOff را نیز بزرگ بگیرید که روی انرژی و چگالی نویز زیادی ایجاد نشود.
انتخاب و بهینهسازی پایهها + واهلش ساختاری
در آموزش این جلسه نحوه انتخاب پایهها به روش Split-Valence و بهینهسازی پارامترهای وابسته معرفی میشود.
به صورت کلی برای بهینهسازی این پایهها و خصوصاً در مورد پایههای DZP دو پارامتر در سیستا در نظر گرفته شده است. PAO.EnergyShift و PAO.SplitNorm که میتوانید با تغییر این دو پارامتر دقیقاً مشابه روشی که در جلسه پیش برای kpoint ها انجام شد، پایهها را تا حد ممکن نسبت به تغییرات انرژی پایدار کنید.
در روند بهینه سازی به این نکات توجه کنید – این کارها باید به ترتیب انجام شوند:
1- انتخاب پایه مناسب برای مثال DZP – TZP – SZ یا TZ3P و … و بهینه سازی شعاع های قطع متناطر با بخش زتای آنها
2- بهینه سازی 1- meshCufOff و 2- kpoint ها یا تعداد نقاط منطقه اول بریلوئن
3- واهلش ساختاری یا ریلکس سامانه جهت پیدا کردن کمینه سطح انرژی پتانسیل (PES) : برای واهلش ساختار میتوانید از هر روشی استفاده کنید. فقط معیارتان را روی همگرا شدن قرار دهید. ممکن است سامانه شما با یک نوع محاسبات به همگرایی نرسد یا درون یک مینمم موضعی قرار گیرد.
ساختار نواری و چگالی حالتهای الکترونی با گنوپلات
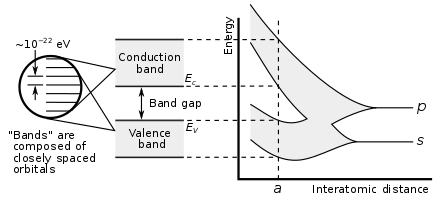
ساختار نواری جامدات همیشه از جذابیت برخوردار بوده است. یکی از مهمترین ابزارهای فیزیک و در حقیقت نقطه قوت فیزیک کوانتوم که جهشی در علم مواد جامد ایجاد کرد، خلق ابزاری بنام ساختار نواری بود. در فیزیک حالت جامد، ساختار نواری الکترونی محدوه ای از انرژی را توصیف میکند که ممکن است الکترون دارای آن انرژی باشد (نوارهای انرژی یا انرژی های مجاز) و محدوده ای که الکترون نمیتواند دارای انرژی باشد ( گاف نواری یا انرژیهای ممنوعه ).
اما سوال اصلی اینست که چنین ایده ای از کجا شروع شد؟
از وقتی متوجه شدیم یک اتم منزوی و ایزوله دارای ترازهای انرژی گسستهای است. برای مثال اتم هیدروژن را در نظر بگیرید: دارای بینهایت تراز انرژی است. اما چندتا از این ترازها دارای الکترون است؟؟؟
تنها یکی! الکترون یک فرمیون است و فرمیونها با آمار فرمی ترازهای انرژی را پر میکنند (برای نمونه قاعده هوند در پر شدن ترازها در درس شیمی را بیاد بیاورید). اگر دما صفر مطلق باشد: Ground state سامانه یا همان حالت زمینه سامانه با رعایت فرمیون بودن پر خواهد شد.
اگر دو اتم هیدروژن را بهم نزدیک کنید، چه اتفاقی می افتد؟
تشکیل یک مولکول دو اتمی را میدهد که دو اوربیتال مولکولی را ایجاد میکند. به یکی از این اوربیتالها، پیوندی و به دیگری پادپیوندی میگویند. اوربیتال پیوندی طبق اصل طرد پائولی میتواند پذیرای دو الکترون باشد که هر الکترون نیز از هیدروژن منزوی میآید. بعلاوه سطح انرژی اوربیتالها در ۳ پیکربندی مختلف دارای اندازههای متفاوت است:
۱- وقتی هر الکترون در اتم منزوی هیدروژن قرار دارد.
۲- وقتی الکترونها در اوربیتال پادپیوندی قرار دارند.
۳- وقتی الکترونها در اوربیتال پیوندی هستند.
منطقی است که کمترین سطح انرژی مربوط به وقتی است که الکترونها در اوربیتال پیوندی باشند، چون این اوربیتال حاصل از تداخل سازندهی اوربیتالهای الکترونی اتمهای منزوی است.
پس در مثال دو اتمی فوق دو تراز جدید از انرژی تشکیل شد که با همتای اتم منزویاش متفاوت بود. یکی از اینها پر از الکترونها ( bonding = پیوندی ) بود و دیگری خالی از الکترونها ( Anti-Bonding = پادپیوندی ) بود. یعنی دقیقا مثل مورد یک اتم منزوی که دارای ترازهای گسستهای از انرژی بود.
ساختار نواری
حالا بیایید همین را به تعداد بسیار زیادی از اتمهای هیدروژن تعمیم دهیم. اگر مثلا به تعداد عدد آووگادرو اتم هیدروژن در اختیار داشته باشیم، که در فاصلهی بینهایت از یکدیگر قرار دارند، وقتی این اتمها را به هم نزدیک میکنیم بهگونهای که بین اوربیتالهای الکترونی اتم منزوی همپوشانی به وجود آید، اوربیتالهای مولکولی جدیدی بوجود میآیند. اما دیگر محدود به دو اوربیتال پیوندی و پاد پیوندی نیستند. بلکه بینهایت جهت گیری و همپوشانی فازی مختلف بین اوبیتالهای اتم منزوی ایجاد خواهد شد. کمانرژیترین اوربیتالی که به وجود میآید همان پیوندی است ولی تعداد زیادی اوربیتال دیگر بین پیوندی و پادپیوندی ایجاد میشود. اما به نظرتان چرا فقط همان دو اوربیتال ایجاد نشد؟؟
طبق اصل طرد پائولی در یک سامانهی فرمیونی با ۲۳^۱۰ فرمیون، این تعداد فرمیون نمیتوانند در یک تراز انرژی با اعداد یکسان کوانتومی جای گیرند. این دلیل خوبی است که یک تراز انرژی در یک اتم منزوی، به تعداد زیادی تراز انرژی (در مورد بلوری از اتمهای هیدروژن) با فاصلهی بسیار اندک شکافته میشود.
نوارهای انرژی اینگونه ایجاد میشوند. نوارهای انرژی مجاز نوارهایی هستند که الکترون میتواند درون آنها بنشیند. در جاهایی هم هیچگونه نواری وجود ندارد، گاف داریم.
تعدادی از نوارهای انرژی پر شده هستند. اما این یک سقف دارد. از جایی به بعد الکترونی وجود ندارد که بخواهد stateها را پر کند. آنجا همان جایی است که معروف به انرژی فرمی است.
انرژی فرمی در عایق ها کاملاً نوارهای پر و خالی را از هم جدا می کند. اما در فلزات با نوارهای باند رسانش تلاقی میکند.
یک مطلب هم در پرانتز: یک تفاوت بین انرژی فرمی و تراز فرمی وجود دارد. انرژی فرمی در فیزیک حالت جامد به دمای صفر اطلاق میشود ولی تراز فرمی در دمای متناهی نیز معتبر است که با آماری همچون توزیع فرمی-دیراک توصیف میشود.
توصیفی که در بالا ظاهر شد، بواسطهی اینکه انرژی بر حسب فاصلهی شبکه بود در فضای مستقیم ارائه شد. اما بلور یک ساختار دورهای است. فضای وارونی نیز دارد که در آن فضا توصیف کمیتها و حتی همین ساختار نواری شکل سادهتری برای توصیف دارد. گرفتن تبدیل فوریه در همهجای علوم مرسوم است و به ساده کردن فهم مطلب و حل راحتتر کمک میکند.
در فیزیک حالت جامد که شاخهی کوچکی از علم ماده چگال است اما، رابطه انرژی بر حسب عدد موج که به رابطه پاشندگی انرژی معروف است میتواند ساختار نواری را به شکل آشناتری تبدیل کند.
تفسیرهای فوقالعاده جالبی میتوان از دل همین ساختار نواری بیرون کشید. برای مثال فرض کنید یک ماده عایق دارید.
در مواد عایق همیشه نوارهای باند والانس به صورت کامل پر هستند و در حقیقت الکترونهای والانس مشخصاً باید تعداد زوجی باشند. درون هر نوار نیز طبق اصل طرد پائولی میتواند الکترونی با اسپین بالا و الکترونی با اسپین پایین یک نوار را در یک k اشغال کنند(غیرمغناطیسی). پس گویا هر نوار به ازای یک k باید متناظر با ۲ الکترون باشد.
درون یاختهی یک ترکیب عایق تعداد الکترونها زوج است. یعنی نوارها به صورت کامل پر شده هستند و با اعمال میدان خارجی هیچ شارش الکترونی به وجود نخواهد آمد.
اما اگر نوار نیمه پر باشد یعنی یاخته فرد تا الکترون دارد و رسانا است.
حالتهای خاصی نیز در طبیعت یافت میشود که تعداد پر شدگی stateهای کان-شم (البته در فرمالیسم DFT) فردتاست، اما ماده کاملا عایق است. این موارد اکنون جز موضوعات داغ هستند. نمونههای مشهور عایق مات و سامانههای همبسته قوی است.
در این جلسه از گنوپلات برای ترسیم ساختار نواری و چگالی حالتهای الکترونی استفاده شده است. نمونه کد استفاده شده در ویدئو:
set term postscript eps enhanced
set output “band.ps”
unset xtics
set label “{/Times-Roman X}” at 0.602324,-8.5
set label “{/Symbol G}” at 1.214647,-8.5
set label “{/Times-Roman L}” at 1.754935,-8.5
set label “{/Symbol G}” at 0,-8.5
set xrange [0:1.7549]
set yrange [-8:4]
set bmargin 2
set arrow 1 from 1.224647,-8 to 1.224647,4 nohead
set arrow 2 from 0.612324,-8 to 0.612324,4 nohead
set arrow 3 from 0,0 to 1.754935,0 nohead
set ylabel “E-E_f(eV)” offset 0,5
plot “Si.dat” us 1:($2+5.103313) w l lt 8 lw 1.5 title “Silicon Band”
set output “band.ps”
unset xtics
set label “{/Times-Roman X}” at 0.602324,-8.5
set label “{/Symbol G}” at 1.214647,-8.5
set label “{/Times-Roman L}” at 1.754935,-8.5
set label “{/Symbol G}” at 0,-8.5
set xrange [0:1.7549]
set yrange [-8:4]
set bmargin 2
set arrow 1 from 1.224647,-8 to 1.224647,4 nohead
set arrow 2 from 0.612324,-8 to 0.612324,4 nohead
set arrow 3 from 0,0 to 1.754935,0 nohead
set ylabel “E-E_f(eV)” offset 0,5
plot “Si.dat” us 1:($2+5.103313) w l lt 8 lw 1.5 title “Silicon Band”
ارتعاشات شبکه و مدهای فونونی
در این آموزش ویدئویی، محاسبات فونونی برای بلور الماسی Si انجام شده و ساختار نواری فونونی آن بدست آمده است. همچنین اطلاعات مربوط به ثابت های نیرو و ویژه مدها نیز در خروجی بدست آمدهاند.
همچنین شما را به پیوند زیر بازگشت میدهم تا با روشهای BRR, 1DMIN که همان روش گفته شده در آموزش است، آشنا شوید:
جذب یکی از موضوعات مطرح در شیمی فیزیک است. در این جلسه یک اتم هیدروژن روی سطح ۰۰۱ آلومینیم و جایگاه top یا onsite نشانده میشود. در جلسات بعدی نیز انرژی جذب و مکان جذب مختلف بررسی میشود.
توجه
برای دسترسی به فصل های این دوره باید ابتدا در دوره ثبت نام کنید.
توجه
برای شروع پرسش و پاسخ باید در دوره ثبت نام کنید !

از همین مدرس
محصولات مشابه



وضعیت دوره
دوره درحال برگزاری هست.
480 هزار تومان
سیستا یا SIESTA یک کد محاسبات کوانتومی است که توانایی انجام محاسبات ابتدا به ساکن الکترونیکی (ab initio) و محاسبات دینامیک مولکولی (molecular dynamics) را دارد.
حداقل ۱ امتیاز ضروری هست
| 60دانشجو |
| 20 درس |
| آخرین آپدیت : جمعه, 19 اردیبهشت 4 |
| سطح دوره : متوسط |
| زبان : فارسی |
| نوع دوره :غیر حضوری |
| زمان دوره :نامعلوم |
| حجم فایل ها : کمتر از ۱ گیگابایت |
| پشتیبانی :تلگرام |
| پیش نیاز :فیزیک پایه |
| بدون دیدگاه |
| 2512 مشاهده |
| برچسب: dft سیستا نظریه تابعی چگالی |
| دسته: فیزیک |
| لینک کوتاه دوره: |
مشاوره رایگان
برای کسب اطلاعات بیشتر درباره این دوره درخواست مشاوره خود را ارسال کنید و یا با ما تماس بگیرید.
دیدگاهها
هیچ دیدگاهی برای این محصول نوشته نشده است.